Protein Folding
After I received my doctoral degree in Mathematics at the Universidad Nacional del Sur in Bahía Blanca, Argentina, I carried out research at the University of Chicago (in the groups of professors Tobin Sosnick, Karl Freed, and Steve Berry) on the so-called Protein Folding Problem, where the goal is to predict, using computational methods, the three-dimensional structure of a protein molecule given its amino-acid sequence:
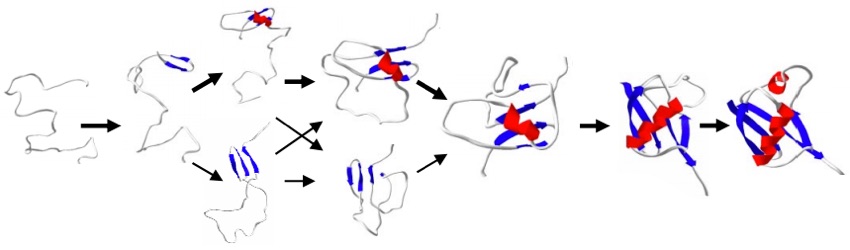
The video below gives a nice non-expert introduction to the protein folding folding problem, and its implications in biotechnology and medicine:
I created algorithms that folded proteins with fewer computational resources than traditional methods by using a simplified, or coarse-grained, representation of the protein molecule:
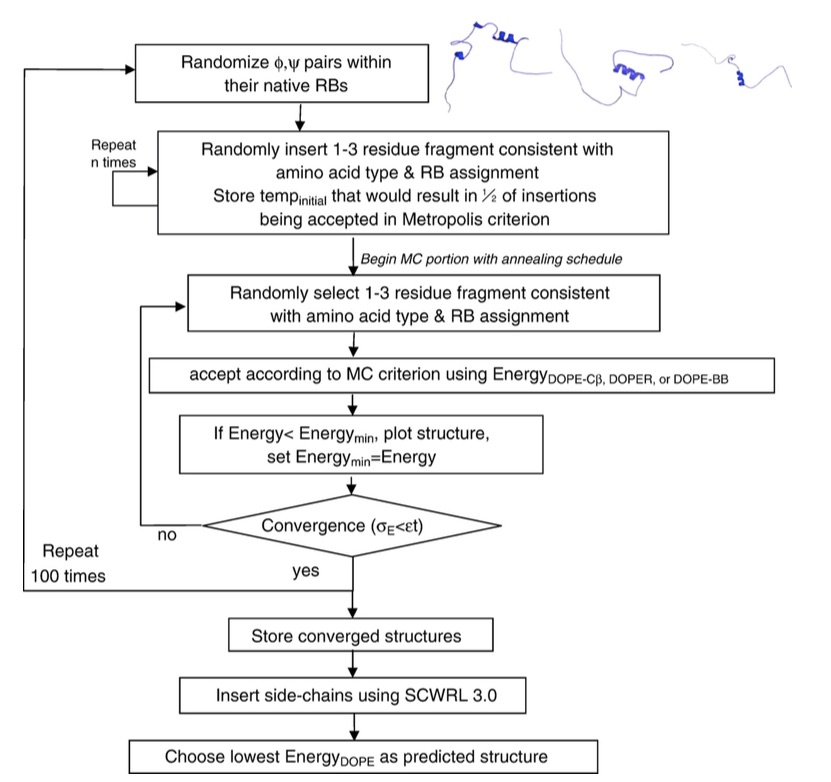
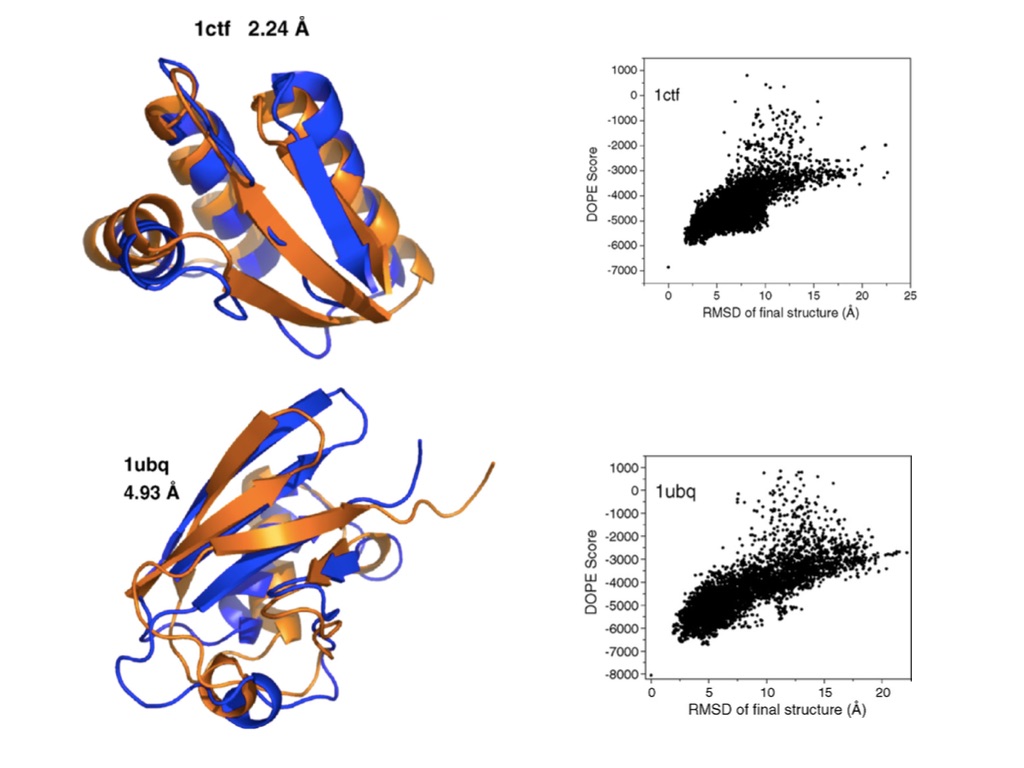
These folding algorithms predicted structures that were fairly close to the real structure of small, globular proteins, as seen in the image below (the ribbon diagrams on the left represent the 3D structure of the predicted and real molecules, and the scatter plots on the right show the “energy” of the simulated structures in the vertical axis versus the distance between the simulated structure and the target structure):
I published a number of academic papers on that subject, such as this one summarizing most of my research at the University of Chicago. All the software tools I developed at that time to run our “coarse-grained” folding algorithms, including the Protein Library, a general and extensible C library and utilities to implement simulations of macromolecules, are available online here.